Long-read Sequencing for Livestock Genomics and Breeding
Long-read Sequencing for Livestock Genomics and Breeding
Blog Article
Overview
Long-read sequencing technology is helping researchers and breeders produce healthier, more productive livestock. Agricultural genomics has and will continue to drive sustainable productivity and provide solutions to the growing challenges facing the global population. With long-read sequencing technology, farmers, breeders, and researchers are able to:
- Attribute desired traits to single nucleotide variants (SNVs), structural variants (SVs), and complex genotypes.
- Capture genomic variation in distant, inbred, and populations on a genome-wide scale.
- Construct reference-quality, haplotype-resolved pan-genomes to drive marker development, trait discovery, and germplasm characterization.
Many livestock studies have utilized sequence-level variation to understand population-scale diversity and genetic improvement of livestock. However, most of these studies are limited to SNVs and small insertions/deletions (INDELs) detected by short-read sequencing. SVs involving longer DNA fragments have not been extensively studied in livestock, particularly at the genome-wide and population scales. Long-read sequencing technology (Pacific Biosciences and Oxford Nanopore sequencing) provides high-precision reads across thousands of base lengths, making it an ideal tool for rapidly assembling phased, reference-quality genomes and addressing some of the shortcomings of short-read long sequencing for SV discovery and genotyping. Currently, long-read sequencing technology is widely used in genomics studies of livestock species.
Importance of Structural Variants in Livestock Breeding
Structural variants, including deletions, insertions, inversions, and copy number variations, have become important genetic markers for understanding genomic diversity. In livestock breeding, these variants provide a special perspective for understanding the genetic determinants that influence economically significant traits. For example, specific SVs are associated with partial or complete knockouts that directly affect phenotypic outcomes. Empirical evidence from a variety of studies (e.g., human, plant, and animal studies) confirms the impact of SVs on genomic function.
Given the large size of SVs, they are more capable of influencing gene function than SNPs. While many SVs found in livestock show single-gene effects on traits, discerning the exact molecular impact remains challenging due to cost constraints and logistical issues associated with accessing genetic material. However, as long-read sequencing technologies become more affordable, pinpointing SVs affecting quantitative traits becomes more feasible. In addition, as our livestock genetic databases grow in depth and breadth, we are getting closer to cataloging a comprehensive list of SVs, thus providing a more comprehensive view of livestock genomics.
Livestock Structural Variation Population-scale Studies Using Long-read Long Sequencing
Developing large long-read sequencing reference populations
One of the strategies of modern genomics is the development of reference populations, which serve as the base dataset for various genetic analyses. Long-read sequencing has unique advantages over short-read sequencing for livestock species. For example, it provides better resolution for SV, allows accurate phasing of longer haplotypes, and ensures superior interpolation accuracy.
Ideally, these reference populations should be diverse, including a large number of individuals from popular breeds, representatives from rare breeds, and even ancestral livestock lineages. By having a comprehensive dataset, researchers can facilitate the discovery and estimation of SVs, thereby advancing livestock breeding.
Validation of the SV effect and its molecular mechanisms
To truly realize the potential of SVs, it is critical to understand their effects and underlying molecular mechanisms. Validation of the discovered SVs can be achieved through a variety of wet-lab methods, including remote PCR amplification and Bionano optical mapping. In addition, familial patterns of SV inheritance can provide insights into their authenticity.
SVs play multiple roles in gene regulation. They can affect promoter and enhancer activity, alter gene expression patterns, and in some cases, even lead to gene fusions or fragmentation. Genome-wide expression quantitative trait loci (eQTL) mapping provides an interesting approach to elucidate the effects of SVs on macro-scale gene expression. Recent studies suggest that SVs may have a more significant impact on gene regulation than previously anticipated, making them valuable genetic markers in livestock breeding.
Utilizing SV knowledge to develop genomic tools in animal breeding
A comprehensive understanding and catalog of SVs provide a foundation for their practical application in livestock breeding. Considerations include the commonality of functional SVs, the accuracy of interpolation, and the potential for integration into various genotyping platforms. Combining SVs with SNVs in genomic prediction models can provide nuanced insights into the inheritance of livestock traits. In addition, technologies like CRISPR gene editing present a promising future for targeting specific SVs to induce desired phenotypic outcomes in livestock. A new bovine genome ARS-UCD1.2 was detected based on PacBio sequencing technology. Thus, a more comprehensive, complete, and precise bovine reference genome has helped scientists redesign their gene-editing protocols. Such targeted breeding practices could revolutionize the livestock industry, ensuring improved animal productivity, health, and welfare.
Examples of Long-read Sequencing for Livestock Breeding
PacBio SMRT sequencing methodology has granted scientists an intricate vantage point into the bovine transcriptomic landscape. Employing this technologically advanced tool, investigators have delved deep into the intricate web of the bovine genome, revealing an abundance of full-length non-chimeric sequences. Further, the elucidation of myriad alternative splicing events and the identification of hitherto unknown long non-coding RNAs (lncRNAs) have served a dual purpose: they have substantially augmented the corpus of extant genetic data and have simultaneously furnished livestock breeders with a granular, nuanced blueprint to guide subsequent breeding endeavors.
Moving beyond the bovine species, an ambitious endeavor was undertaken to construct a comprehensive pangenome of the porcine species. This monumental effort, which assimilated genomic data from no fewer than 11 distinct genomes, bore fruit in the form of a veritable compendium of novel sequences and structural variants (SVs). A deeper foray into this plethora of genetic information has unmasked particular SVs that are unique to specific pig breeds. Such insights have been pivotal in accentuating the evolutionary adaptations and idiosyncratic traits endemic to these breeds, thereby enabling scientists and breeders alike to leverage this genetic treasure for targeted breeding and research undertakings.
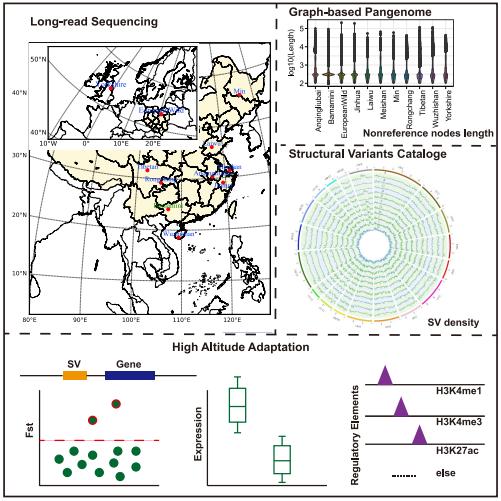
Pangenome obtained by long-read sequencing of 11 genomes reveal hidden functional structural variants in pigs. (Jiang et al., 2023)
References
- Jiang, Yi-Fan, et al. "Pangenome obtained by long-read sequencing of 11 genomes reveal hidden functional structural variants in pigs." Iscience 26.3 (2023).
- Chang, Tianpeng, et al. "PacBio single-molecule long-read sequencing provides new light on the complexity of full-length transcripts in cattle." Frontiers in Genetics 12 (2021): 664974.